Paciente de sexo femenino de 8 años, con antecedentes
de epistaxis anterior recurrente desde
los 2 años, que consulta a nuestro servicio por
telangiectasias faciales de 4 años de evolución,
las que habían ido aumentando de forma progresiva.
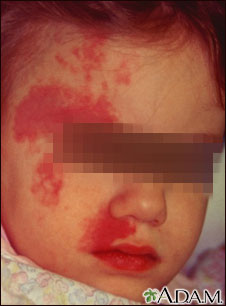
Al examen físico se evidenciaron múltiples
telangiectasias lineales, aracneiformes y
puntiformes en la frente, ambas mejillas (Fig. 1 y
2), cuello, puntas de dedos (Fig. 3), además de
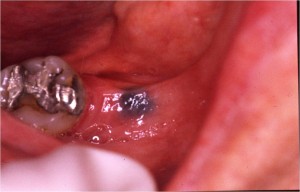
múltiples angiomas puntiformes en los labios y
mucosa labial (Fig. 4). Estas lesiones fueron también
identificadas en el examen físico del padre.
Al interrogarla en forma dirigida, la madre refirió
que la paciente presentaba cefaleas periorbitarias
intensas, aproximadamente 3 veces a la
semana.Por este motivo se le solicitó una angioresonancia
de cerebro que demostró la presencia
de un shunt arteriovenoso intracerebral del
sistema vertebrobasilarLa paciente fue derivada al Instituto de
Neurocirugía, donde fue exitosamente intervenida
con embolización de la malformación con
coils. Posteriormente se solicitaron como parte
del estudio ecografía abdominal, endoscopia
digestiva alta, radiografía de tórax y angioTAC
de tórax, las que resultaron normales.
Actualmente la paciente se encuentra en control
con un equipo multidisciplinario, en buenas
condiciones y completando su estudio en busca
de otras MAV.
atte, nahim I Valenzo